
When you pick up a generic drug at the pharmacy, you expect it to work just like the brand-name version. That expectation isn’t luck-it’s the result of strict science and regulation. The U.S. Food and Drug Administration (FDA) doesn’t just approve generic drugs because they look the same or cost less. They require manufacturers to prove bioequivalence-that the generic delivers the same amount of active ingredient, at the same speed, into your bloodstream as the original drug. Without this proof, a generic drug can’t be sold in the U.S.
What Bioequivalence Really Means
Bioequivalence isn’t about how the drug tastes or what color the pill is. It’s about what happens inside your body. The FDA defines it as: "the absence of a significant difference in the rate and extent to which the active ingredient becomes available at the site of drug action." In plain terms, if you take a generic drug and the brand-name version, your body should absorb and use them in nearly identical ways.This matters because even small differences in absorption can affect how well a drug works-or whether it causes side effects. For example, a drug like warfarin, used to prevent blood clots, has a very narrow window between being effective and being dangerous. If a generic version absorbed 20% slower, it could mean the difference between preventing a stroke and causing a bleed.
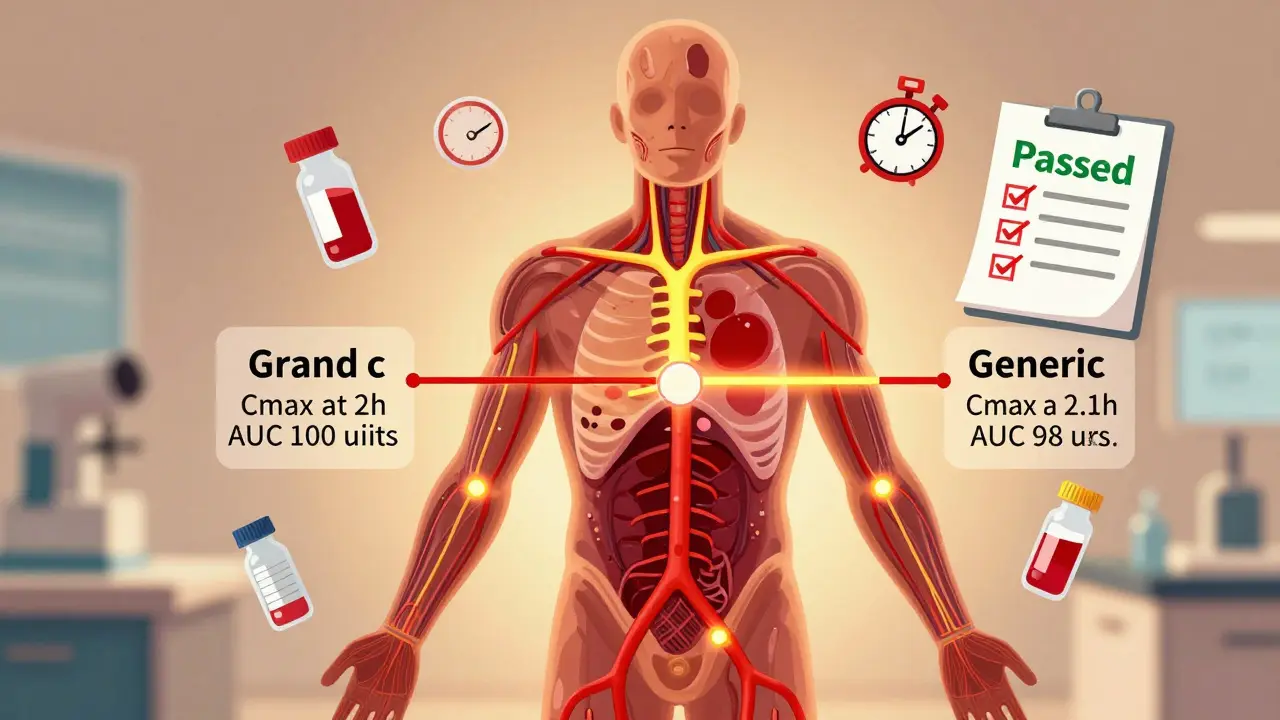
The 80/125 Rule: The Gold Standard
To prove bioequivalence, manufacturers run clinical studies in healthy volunteers. These studies measure two key numbers:- AUC (Area Under the Curve): How much of the drug enters your bloodstream over time (total exposure).
- Cmax (Maximum Concentration): How fast the drug reaches its peak level in your blood.
The FDA requires that the 90% confidence interval for the ratio of these values-test (generic) to reference (brand)-must fall between 80% and 125%. This is known as the 80/125 rule. It’s been the standard since 1992 and hasn’t changed.
For example, if the brand-name drug gives an AUC of 100 units, the generic must deliver between 80 and 125 units. If it’s 79 or 126, the study fails. This isn’t arbitrary-it’s based on decades of data showing that drugs within this range have the same therapeutic effect and safety profile.
How the Studies Are Done
A typical bioequivalence study involves 24 to 36 healthy adults. They’re given either the generic or the brand-name drug, then have their blood drawn over several hours to track drug levels. After a washout period, they take the other version. This is called a crossover design.Most studies are done under fasting conditions-no food for at least 10 hours before dosing. But if the drug is affected by food (like some antibiotics or cholesterol meds), a second study is required under fed conditions. The FDA also requires strict controls: samples must be handled, stored, and analyzed using validated methods that meet Good Laboratory Practice (GLP) standards. Any slip-up in sample handling can invalidate the whole study.
When Bioequivalence Studies Aren’t Needed
Not every generic drug needs a full clinical trial. The FDA allows biowaivers for certain products where absorption is predictable. These include:- Oral solutions with the same active and inactive ingredients as an approved product.
- Topical products meant to work locally (like steroid creams or antifungal ointments), where systemic absorption doesn’t matter.
- Inhalant anesthetics that are delivered as gases.
To qualify for a biowaiver, the generic must meet the Q1-Q2-Q3 criteria:
- Q1: Same active and inactive ingredients.
- Q2: Same dosage form and strength.
- Q3: Same pH, solubility, and dissolution profile as the brand.
For topical products, manufacturers can use in vitro release testing (IVRT) and in vitro permeation testing (IVPT) instead of human trials. These tests measure how quickly the drug comes out of the cream and how well it passes through skin layers. If the results match the brand, the FDA may approve without human studies.
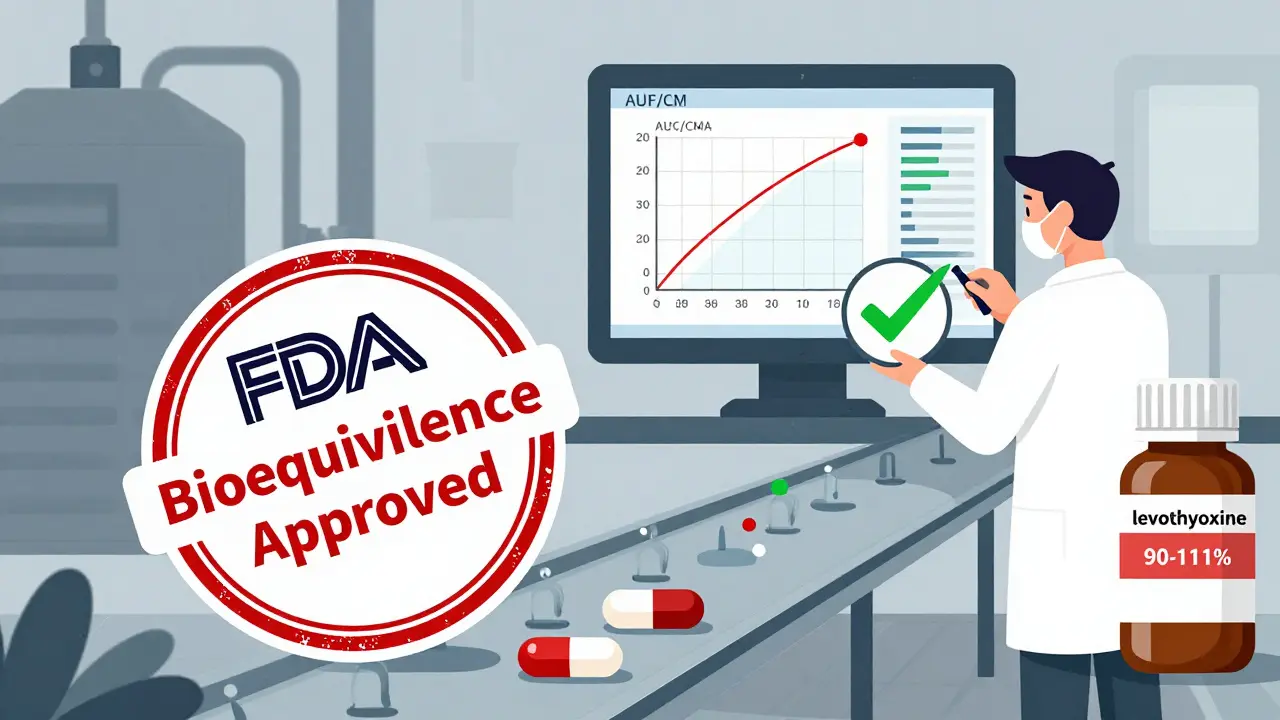
Special Cases: Narrow Therapeutic Index Drugs
Some drugs have such a tight safety margin that the 80/125 rule isn’t enough. For drugs like levothyroxine (for thyroid), warfarin, or cyclosporine, the FDA uses a stricter range: 90% to 111%. This means the generic can’t be more than 11% different from the brand in absorption.These are called Narrow Therapeutic Index Drugs (NTIDs). Even a 5% difference can lead to treatment failure or toxicity. The FDA requires more complex study designs and sometimes multiple bioequivalence studies to ensure safety. Manufacturers of NTID generics face higher scrutiny and longer review times.
Why Many Applications Get Rejected
In 2022, only 43% of generic drug applications (ANDAs) got approved on the first try. The most common reasons? Poor study design, small sample sizes, unreliable lab methods, and incomplete documentation. One company submitted a study that used outdated blood sampling times-missing the true peak concentration. The FDA rejected it.Companies that follow the FDA’s Product-Specific Guidance (PSG) documents have a 68% first-cycle approval rate. Those who don’t? Just 29%. These guidances are tailored for each drug-covering everything from dissolution testing to statistical methods. Ignoring them is like building a house without blueprints.
The Cost and Time of Proof
Running a single bioequivalence study costs between $500,000 and $2 million. That’s why many manufacturers try to use biowaivers or in vitro methods where possible. For a company making 10 generics, that’s $5 million to $20 million just in testing costs.The FDA’s Domestic Generic Drug Manufacturing Pilot Program helps by speeding up review for generics made in the U.S. with U.S.-sourced ingredients. But even with faster review, the entire ANDA process still takes 14 to 18 months on average. Bioequivalence data is the biggest bottleneck.
The Bigger Picture
Generic drugs make up 90% of all prescriptions filled in the U.S. but only 23% of total drug spending. That’s billions in savings every year. But none of that would be possible without bioequivalence studies. They’re the invisible bridge between innovation and affordability.The FDA is now exploring new tools like physiologically based pharmacokinetic (PBPK) modeling-computer simulations that predict how a drug behaves in the body. For complex products like inhalers or topical gels, these models may one day replace some human trials. But for now, blood samples, controlled conditions, and strict statistical limits remain the gold standard.
What happens if a generic drug fails bioequivalence testing?
If a generic fails bioequivalence testing, the FDA issues a Complete Response Letter (CRL) outlining the deficiencies. The manufacturer must fix the issues-often by redesigning the formulation, repeating the study, or providing additional data. They can resubmit, but each cycle adds months to the approval timeline. Many companies abandon the application after one failure due to cost.
Can a generic drug be approved without human trials?
Yes, but only for certain products. Biowaivers are granted for simple oral solutions, topical products for local effect, and some inhalants if they meet Q1-Q2-Q3 criteria and demonstrate equivalent in vitro performance. For most systemic drugs, especially those taken as pills or capsules, human clinical studies are still required.
Why does the FDA require fasting conditions for bioequivalence studies?
Fasting removes variables like food type, fat content, and digestion speed that can alter how a drug is absorbed. This gives the clearest picture of the drug’s inherent absorption profile. If food affects absorption, a second fed study is required to ensure the generic works the same way under real-world conditions.
Are bioequivalence standards the same in Europe and the U.S.?
Yes, mostly. The FDA and the European Medicines Agency (EMA) both use the 80/125% range for most drugs. Over 87% of their requirements are aligned thanks to international collaboration through the ICH. Differences mostly appear in specific guidance for complex products, like transdermal patches or long-acting injectables.
How do manufacturers choose which reference drug to compare against?
Manufacturers must use the FDA’s Reference Listed Drug (RLD), which is the original brand-name product approved under a New Drug Application (NDA). The RLD is listed in the FDA’s Orange Book. You can’t pick a different brand or a foreign version. Using the wrong RLD is one of the most common submission errors and leads to automatic rejection.
Kyle Young
March 20, 2026 AT 04:22It's fascinating how such a rigid statistical framework-80% to 125%-can govern something as intimate as how our bodies absorb medicine. It makes you wonder: if we can quantify absorption so precisely, why haven't we applied similar rigor to mental health treatments or nutritional supplements? The science exists, but the regulatory will doesn't. There's a philosophical gap here between what we *can* measure and what we *choose* to regulate.
lawanna major
March 21, 2026 AT 00:13I appreciate how the FDA maintains consistency across drugs, especially with NTIDs. It’s not just about cost savings-it’s about trust. When someone takes a generic, they shouldn’t have to second-guess whether their life-saving medication will work. That quiet reliability is a public health triumph, even if no one notices it.
Linda Olsson
March 21, 2026 AT 06:09Let’s be real-this whole bioequivalence system is a controlled illusion. The studies are done on healthy young adults, but most patients are elderly, diabetic, or on five other meds. The ‘80/125 rule’? A marketing loophole disguised as science. And don’t get me started on how the same company often owns both the brand and the generic. It’s all a rigged game.
Ayan Khan
March 22, 2026 AT 14:29In India, we see this daily-generics save lives, but the quality varies wildly because enforcement isn’t uniform. The U.S. system, flawed as it is, sets a global standard. It’s not perfect, but it’s a model. We need more countries to adopt this level of rigor, not less. The science behind bioequivalence is one of the few things in pharma that actually prioritizes patient safety over profit.
Emily Hager
March 23, 2026 AT 13:59I find it deeply troubling that the FDA allows biowaivers for topical products. How can we possibly guarantee equivalence without human trials? This is the same logic that led to the opioid crisis-relying on theoretical models instead of real-world data. We’re trading safety for speed, and someone will pay the price.
Lauren Volpi
March 23, 2026 AT 16:30So let me get this straight. We spend millions to prove a pill works the same… but we let Big Pharma charge $500,000 for a cancer drug? This whole system is a scam. The ‘savings’ are a lie. The real savings go to shareholders, not patients. And don’t even get me started on how they ‘reformulate’ generics to skirt the rules.
Kal Lambert
March 23, 2026 AT 22:50Biowaivers are smart. They cut costs without cutting safety. If the chemistry and dissolution match, why test 30 people? It’s waste. The 80/125 rule works because it’s based on real data, not guesswork. Let the science lead.
Melissa Stansbury
March 25, 2026 AT 16:55Wait, so if a generic fails, they have to redo the whole study? That’s insane. What if they just tweak the coating or the filler? Isn’t there a way to adjust without starting over? Why not let them submit a minor change and re-test just that part? This feels like bureaucratic overkill.
cara s
March 26, 2026 AT 19:08It’s ironic, really. We demand near-perfect bioequivalence for pills we swallow, yet we have zero oversight on vitamins, herbal supplements, or even CBD products. The FDA’s rigor here is a beacon of science… in a sea of regulatory chaos. It makes you wonder what else we’re blindly trusting while ignoring the obvious.
Amadi Kenneth
March 27, 2026 AT 12:14They say the FDA is strict-but have you seen how many generics come from China? How do we know the labs aren’t faking data? The blood samples? The storage? The ‘validated methods’? It’s all a facade. I’ve seen reports-labs in Guangdong are using expired reagents. This whole system is a Ponzi scheme. The government knows. They just don’t care.
Shameer Ahammad
March 29, 2026 AT 12:08Let me clarify: the 80/125 rule is not a scientific standard-it is a political compromise. The pharmaceutical industry lobbied for this range because stricter limits would have killed the generic market. The FDA capitulated. This is not science; it is economics masquerading as pharmacology. Anyone who believes this is rigorous is either naive or complicit.
Alexander Pitt
March 29, 2026 AT 13:38The real hero here is the lab technician who handles the blood samples with sterile precision, 18 hours straight, in a quiet room, while no one else notices. This whole system runs on unseen, uncelebrated work. The science is solid because the people behind it refuse to cut corners.